
ResearchZintl-ion clusters
|
 |
In collaboration with Prof. Zhong-Ming Sun (Nankai University,
China), we are exploring the relationship
between structure, bonding and cluster growth
in anionic Zintl clusters. These clusters
typically contain a mixture of transition
metals and tetrel elements (Ge, Sn, Pb), and
the degree to which the elements segregate
offers important insights into alloying
processes. Our latest work in this area has looked at the nature of Fe-Fe interactions in Sn/Pb clusters containg three or four Fe centers. The particular structural motifs, [Fe3Sn18]4- and [Fe4Sn/Pb18]4-
have precedent in either Pd or Cu chemistry, where the metal ions have d10 configurations, so the comparison of structure and magnetism offers an opportunity to explore the impact of a partially filled d shell.
Earlier papers in Angewandte
Chemie explored the nature of mixed
valency in the formally InIII7InI
cluster [Sb@In8Sb12]5-,
and showed that the crystallographic disorder
could be understood in terms of pairwise
delocalisation over InII2
units. A 2020 Nature Comm.
paper reports a family of new mixed gold/lead
clusters. Gold and lead are notoriously
immiscible, and this appears to be reflected
in the discrete clusters, which are based on a
Aux core surrounded by lead
icosahedra.
Our work in this area was summarised in a recent review article co-authored with Professor Stefanie Dehnen and Professor Florian Weigend.
Electronic structure and bonding in
endohedral Zintl clusters
J. E. McGrady, F. Weigend and S. Dehnen Chem. Soc. Rev., 2022, 51,628.
[Cu4@E18]4-
(E = Sn, Pb): Fused Derivatives of
Endohedral Stannaspherene and Plumbaspherene.
L. Qiao, C. Zhang, C.-C. Shu, H. W. T. Morgan,
J. E. McGrady and Z.-M. Sun, J. Am. Chem.
Soc., 2020, 142, 31, 13288–13293:
http://dx.doi.org/10.1021/jacs.0c04815
A
family of lead clusters with precious metal
cores, C.-C. Shu, H.W.T. Morgan. L.
Qiao, J.E. McGrady and Z.-M. Sun, Nature
Comm., 2020, 11,
3477.
Featured as an editor's highlight:
Structure and Bonding in [Sb@In8Sb12]3- and [Sb@In8Sb12]5-, C. Liu, N.V. Tkachenko, I.A. Popov, N. Fedik, X. Min, C.Q. Xu, J. Li, J.E. McGrady, A.I. Boldyrev and Z.-M. Sun, Angew. Chem. Int. Ed., 2019, 58, 8367.
Endohedral Clusters
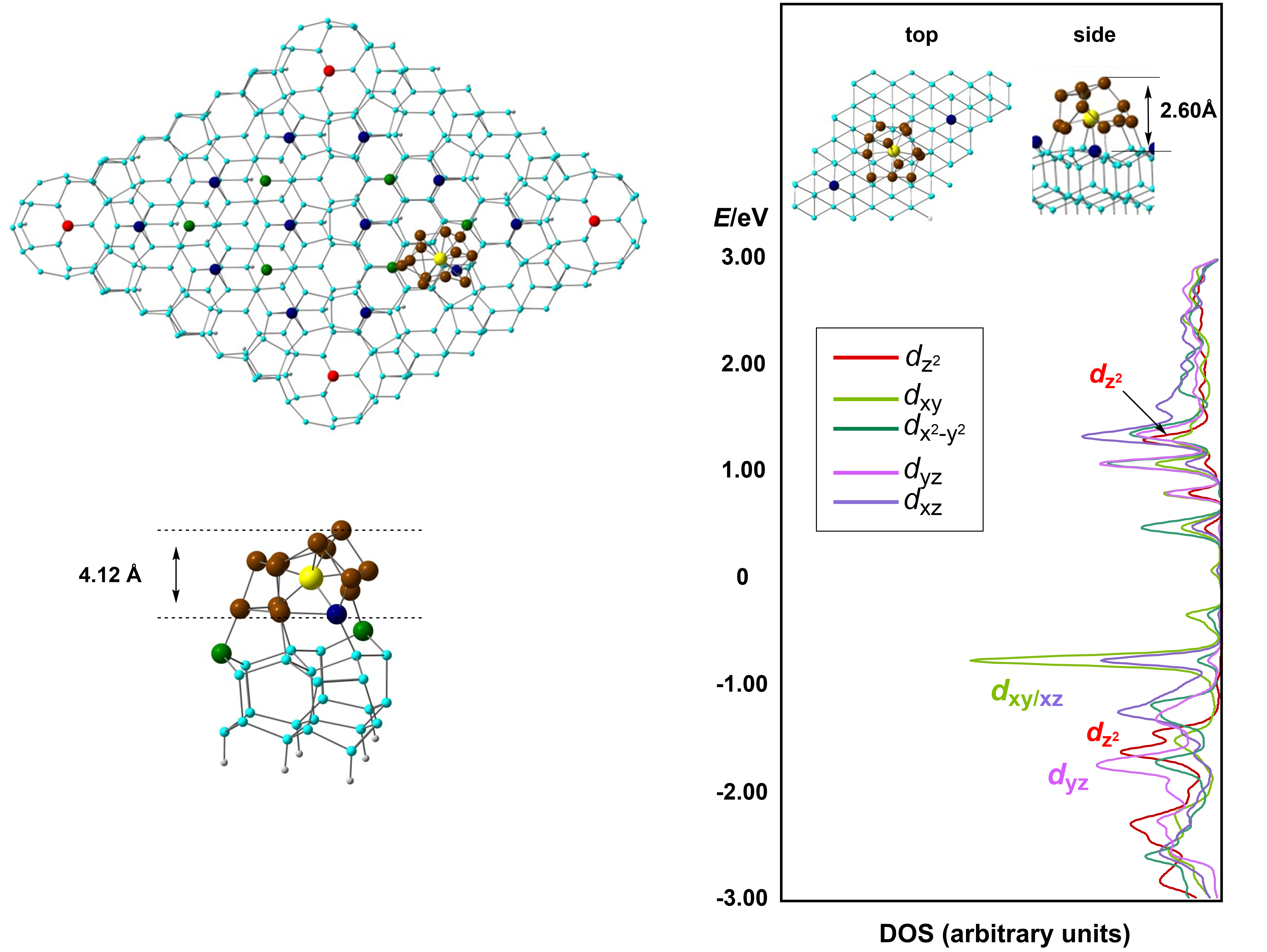 |
As a complement to the
Zintl-ion work in the previous box, we have a
long-standing interest in the electronic
structure of small clusters of Si and Ge which
contain endohedral transition metal ions. This
family is remarkably diverse, and examples are
known for almost all of the transition
elements. These are typically characterised
only in the gas phase through various
spectroscopies, and theory has an important
role to play in establishing structure. More
importantly from our perspective, the nature
of the interaction between the cage and the
metal can vary enormously, and in fact samples
the entire spectrum of chemical bonding. We
have used a range of theoretical tools to
probe these systems, including DFT and
multi-configurational ab initio
methods (CASSCF). Establishing the link
between structure and electronic properties
offers a unique insight into periodic trends.
We have also considered how these interactions
change when clusters are absorbed onto a
silicon surface - this has some relevance to
the behaviour of impurities in bulk silicon, a
matter of great industrial significance.
Low-valent metal
oxides
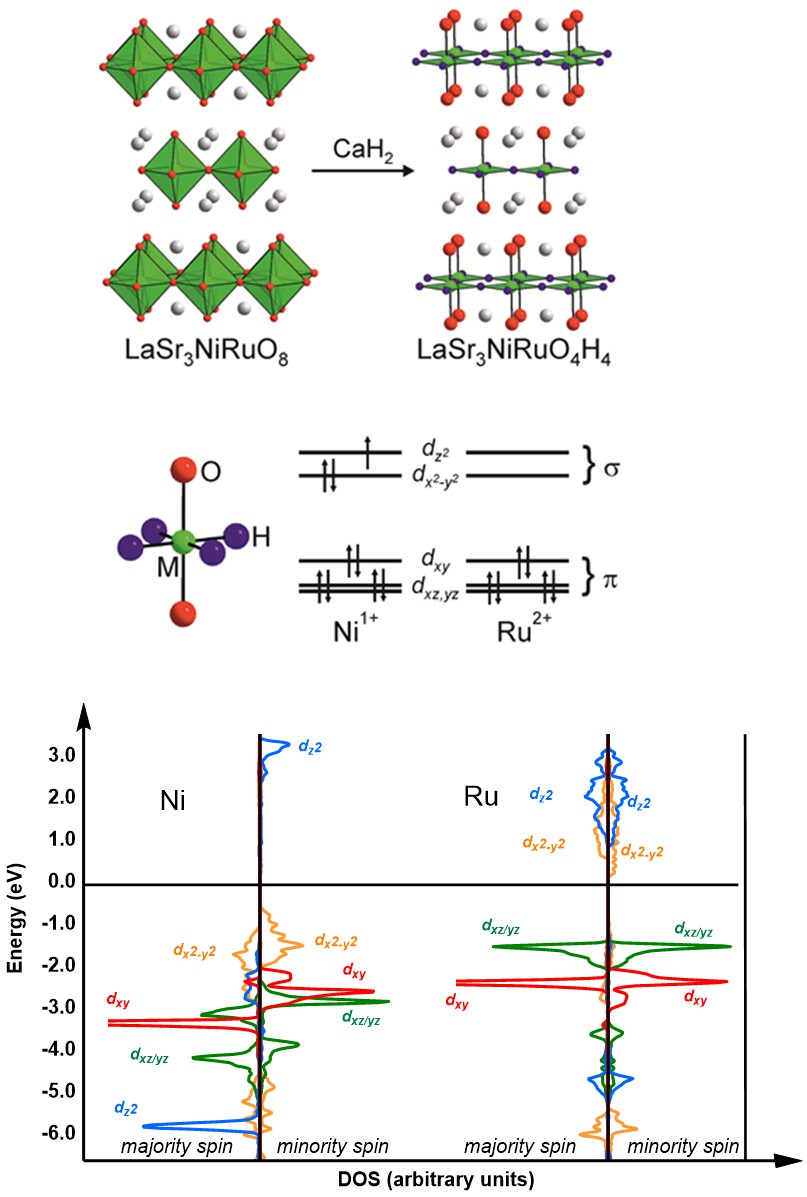 |
Much of our work in the field
of low-valent metal oxide chemistry is done in
collaboration with Professor Michael Hayward,
whose group synthesis novel materials through
topotactic reduction of perovskite-type
materials. The result is either replacement of
oxide with hydride, or complete removal of
layers of oxides, leaving 2-dimensional
materials. The transition metals ions are then
left in unusually low oxidation states (for an
oxide lattice) and, often, also unusual square
planar coordination environments. Our work
involves the application of plane-wave density
functional theory (VASP) along with post
analysis tools such as LOBSTER to analyse
magnetic and electronic properties. In one
recent case study, also in collaboration with
Professor Hiroshi Kageyama, we showed that the
pressure dependence of conductivity in a a
V(III) oxyhydride, SrVO2H, arises
through compression in the xy (VO2)
plane, despite the fact the crystals is
intrinsically more compressible along the z
(H-V-H) axis.
The role of π-blocking hydride ligands in a pressure-induced insulator-to-metal phase transition in SrVO2H, T. Yamamoto, D. Zeng, T. Kawakami, V. Arcisauskaite, K. Yata, M. Amano Patino, N. Izumo, J.E. McGrady, H. Kageyama and M.A. Hayward, Nature Comm., 2017, 8, 1.
Electron transport properties of molecules
 |
Another area of interest to the group is to the link between electronic structure and electron transport properties of extended metal atom chain (EMAC) complexes, where hlical array of oligo-α-pyridyl ligands is used to support a chain of metal centres. These systems have been the subject of a protracted debate in the inorganic chemistry community due to their polymorphism. They exist in symmetric and unsymmetic forms. Our current objective is to relate the fundamental electronic structure of these EMAC complexes to their conductance as measured, for example, by STM. Ultimately, an understanding of these phenomena will be essential to the development of new computer architectures based on molecular-scale components.
Can heterometallic 1-dimensional chains support current rectification?, D. DeBrincat, O. Keers and J. E. McGrady, Chem. Comm., 2013, 49, 9116.
Mechanisms
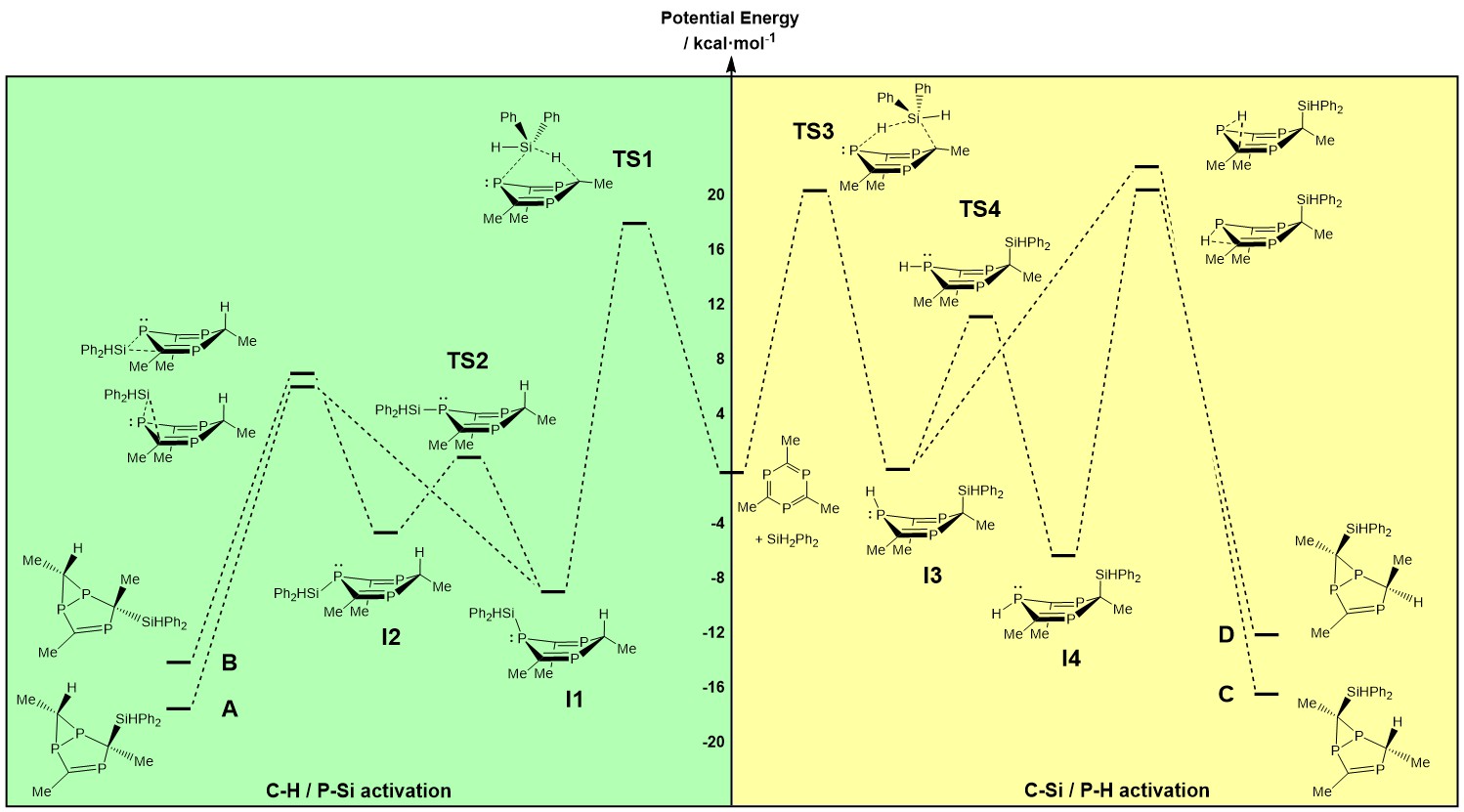 |
We have a number of
collaborative projects that focus on the
mechanisms of chemical reactions. These
involve either transition metal based systems
or main-group reactivity, and our goal is to
identify the fundamental electronic properties
that lead to low barriers. Most recently, in collaboration with the Mehta group in Oxford we have explored the reaction of CO2 with boranes, catalysed by a boron-functionalised P7 cluster.
The goal here is to establish whether the making and breaking of P-P bonds, a well-established phenomenon in the parent P73- cluster, plays a role in the reaction.
Earlier work in collaboration with Chris Russell at Bristol looked at
H-H and Si-H bond activation by the
triphosphabenzene (tBuC)3P3
highlights the degree to which main group
compounds can mimic reactivity (oxidative
additions) typically associated with
transition elements. A very recent new area in
this field is the exploration of metal
clusters on surfaces, and their ability to
catalyse the activation of small molecules.
Hydrogen
Activation by an Aromatic Triphosphabenzene,
L. E. Longobardi, C. A. Russell, M. Green, N.
S. Townsend, K. Wang, A. J. Holmes, S. B.
Duckett, J. E. McGrady and D. W. Stephan, J.
Am. Chem. Soc., 2014, 136,
13453-13457.
Oxidative Addition,
Transmetalation, and Reductive Elimination
at a 2,2'-Bipyridyl-Ligated Gold Center,
M. Harper, C.J. Arthur, J. Crosby, E.J.
Emmett, R.L. Falconer, A.J. Fensham-Smith,
P.J. Gates, T. Leman, J.E. McGrady, J.F. Bower
and C.A. Russell, J. Am. Chem. Soc.,
2018, 140, 4440.
Physical and Theoretical Chemistry Laboratory, South Parks Road, University of Oxford, OX1 3QR, United Kingdom.